【儀表網(wǎng) 研發(fā)快訊】近日,上海交通大學(xué)環(huán)境科學(xué)與工程學(xué)院張禮知教授團(tuán)隊(duì)在美國(guó)化學(xué)會(huì)志《Journal of the American Chemical Society》在線發(fā)表了題為“Regulation of Rh Single-Atom Coordination for Enhanced Reverse Hydrogen Spillover and Efficient Electrochemical Dechlorination”的研究成果。該研究通過調(diào)控鈦基銠單原子電極的配位環(huán)境與電子結(jié)構(gòu),促進(jìn)了原子氫物種從氧化鈦基底向銠單原子活性中心的反向氫溢流過程,以此實(shí)現(xiàn)氯酚類污染物的高效去除。論文第一作者為上海交通大學(xué)環(huán)境科學(xué)與工程學(xué)院博士研究生鄭謙、清華大學(xué)化學(xué)系博士研究生徐恒越,通訊作者為深圳大學(xué)化學(xué)與環(huán)境工程學(xué)院楊波教授、上海交通大學(xué)環(huán)境科學(xué)與工程學(xué)院么艷彩副教授以及張禮知教授,第一完成和通訊單位均為上海交通大學(xué)。
研究背景
反向氫溢流(RHS)作為電化學(xué)加氫反應(yīng)中的關(guān)鍵步驟,在能源存儲(chǔ)、化學(xué)合成及環(huán)境修復(fù)等領(lǐng)域展現(xiàn)出變革性的潛力。傳統(tǒng)的RHS電催化劑通常由具備優(yōu)異水解離能力的可還原金屬氧化物載體(如 TiO2、WO3)和發(fā)生催化反應(yīng)的金屬納米顆粒活性中心組成。其通過在低過電位下產(chǎn)生還原性原子氫物種(H*),并定向傳遞至催化活性中心以完成目標(biāo)反應(yīng)過程(圖1a)。
近年來,單原子RHS電催化劑的興起標(biāo)志著該領(lǐng)域迎來重要范式轉(zhuǎn)變。單原子RHS電催化劑將氫溢流距離縮短至原子尺度(< 1 nm),從而顯著降低氫溢流過程中的反應(yīng)能壘,實(shí)現(xiàn)極高的催化活性和反應(yīng)位點(diǎn)利用效率(圖1b)。盡管前景廣闊,該方向仍存在一定的認(rèn)知空白。尤其對(duì)于單原子位點(diǎn)的配位環(huán)境與電子結(jié)構(gòu)如何調(diào)控其反向氫溢流活性,目前尚缺乏系統(tǒng)深入的理解。揭示其背后的構(gòu)效關(guān)系,將為設(shè)計(jì)下一代高活性、高反應(yīng)選擇性的單原子RHS電催化劑奠定基礎(chǔ)。

圖1 金屬納米顆粒和單原子RHS電催化劑的反應(yīng)路徑比較
研究?jī)?nèi)容
本工作在前期單原子反向氫溢流的研究基礎(chǔ)上(Angew. Chem. Int. Ed. 2024, 63, e202401386),通過調(diào)控煅燒溫度和氣氛,制備了具有不同Rh-O配位環(huán)境的鈦基Rh單原子電極材料(記為Rh1Ox,x = 3、4、5)。
首先對(duì)鈦基Rh單原子電極上的RHS過程開展理論計(jì)算研究(圖2)。Bader 電荷分析顯示,隨著Rh-O配位數(shù)的上升,更多的電子從Rh單原子轉(zhuǎn)移到氧化鈦基底,這種電子損失顯著削弱了Rh與H*之間的電子相互作用。氫吸附吉布斯自由能結(jié)果顯示:Rh1O3構(gòu)型中,Rh 位點(diǎn)對(duì)H*的吸附過強(qiáng)(–0.99 eV),不利于H* 的脫附以及后續(xù)的加氫反應(yīng);在Rh1O5 構(gòu)型中,Rh位點(diǎn)(+0.47 eV)與鄰近O位點(diǎn)(–0.35 eV)間吸附能差異過于顯著,導(dǎo)致極高的溢流能壘,阻礙了 H* 的遷移;相較之下,Rh1O4 構(gòu)型中,Rh位點(diǎn)表現(xiàn)出適中的氫吸附能(+0.08 eV),既滿足H*脫附的熱力學(xué)要求,又顯著降低了氫溢流能壘,從而使其成為潛在最優(yōu)的RHS電催化劑。
態(tài)密度分析顯示,隨著Rh-O配位數(shù)的上升,Rh的d帶中心從 –1.43 eV(Rh1O3)逐漸下移至 –1.66 eV(Rh1O4)和 –1.78 eV(Rh1O5),表明Rh上H*吸附逐級(jí)減弱,與氫吸附能結(jié)果一致。上述分析表明,Rh-O配位數(shù)上升會(huì)導(dǎo)致 Rh d 軌道電子密度下降,進(jìn)而削弱其與H*的相互作用。Rh1O4 獨(dú)特的電子結(jié)構(gòu)、配位構(gòu)型使得H*可以高效地從相鄰O向Rh遷移,并最終脫附、參與還原反應(yīng)。

圖2鈦基Rh單原子電極RHS的理論研究
受理論研究的啟發(fā),對(duì)不同煅燒溫度、氣氛下制備的鈦基Rh單原子電極進(jìn)行材料表征(圖3)。球差電鏡(HAADF-STEM)中孤立的亮點(diǎn)證明Rh物種的原子級(jí)分散。傅里葉變換EXAFS 光譜中顯著的Rh–O 信號(hào)(1.58 Å)進(jìn)一步證實(shí)Rh以單原子形式存在。EXAFS 擬合結(jié)果顯示,Rh1O3、Rh1O4 和 Rh1O5中Rh-O的配位數(shù)分別為 3、4 和 5,與其命名和計(jì)算研究結(jié)果相一致。對(duì) Ti 2p XPS 分析證實(shí)了電極表面氧化鈦的存在,而 Rh 3d XPS光譜則揭示了配位環(huán)境調(diào)控過程中Rh化學(xué)價(jià)態(tài)的改變。在 Rh1O3構(gòu)型中,Rh?(307.5 eV)占主導(dǎo)地位;在 Rh1O3構(gòu)型中,Rh?(308.1 eV)占主導(dǎo)地位;而在 Rh1O5構(gòu)型中,Rh³?(309.5 eV)占主導(dǎo)地位。這種化學(xué)價(jià)態(tài)的變化與巴德電荷的趨勢(shì)一致,明確地將單原子配位環(huán)境與電子結(jié)構(gòu)聯(lián)系起來。

圖3 鈦基Rh單原子電極的材料表征
在此基礎(chǔ)上,利用一系列原位電化學(xué)表征系統(tǒng)地研究了鈦基Rh單原子電極上的反向氫溢流過程(圖4)。氫脫附循環(huán)伏安(CV)結(jié)果顯示,Rh1O4上的H*脫附速率較Rh1O3和Rh1O5顯著加快,證明氧化鈦基底上H*可以通過轉(zhuǎn)移到Rh單原子位點(diǎn),實(shí)現(xiàn)高效的脫附過程。氫吸附電化學(xué)阻抗譜(EIS)結(jié)果顯示,Rh1O4的氫吸附量、氫吸附速率較Rh1O3和Rh1O5均有顯著上升,進(jìn)一步證明是Rh1O4最優(yōu)的RHS電催化劑。原位拉曼光譜結(jié)果顯示,Rh1Ox的Rh-H 伸縮振動(dòng)信號(hào)(1920 cm?¹)在不同的電位下出現(xiàn):Rh1O3為 –1.2 V,Rh1O4為 –0.1 V,而Rh1O5則為 –0.5 V。Rh1O4 在最低的過電位下激活 Rh-H鍵,因而能夠在最溫和的反應(yīng)條件下觸發(fā)反向氫溢流現(xiàn)象。
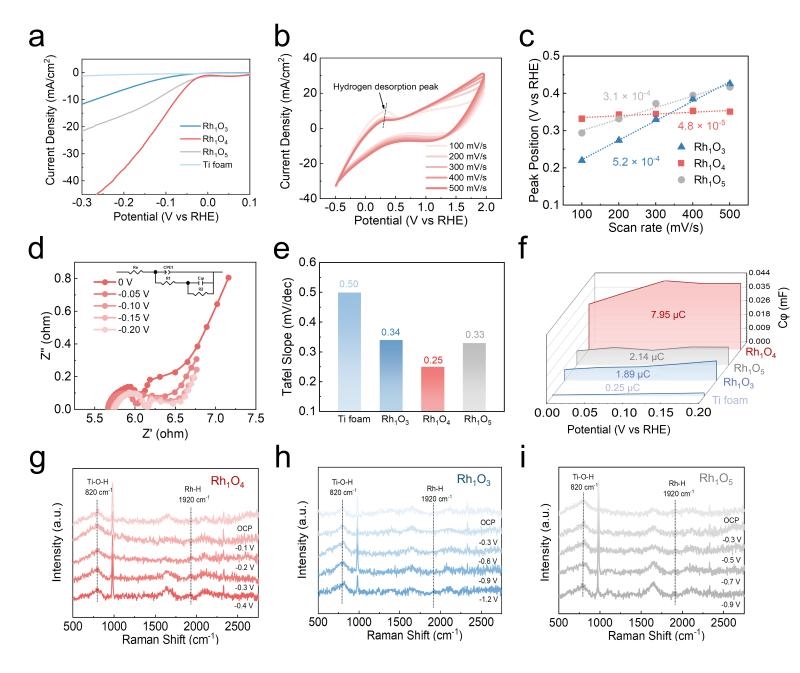
圖4 鈦基Rh單原子電極RHS的原位電化學(xué)表征
使用對(duì)氯苯酚(4-CP)作為模型污染物,測(cè)試了Rh1O4及其對(duì)照樣的電化學(xué)脫氯性能(圖5)。在 -0.6 V vs Hg/HgO的電位條件下,Rh1O4的4-CP還原比例在45 min內(nèi)達(dá)到99%以上,超越Rh1O3(1h,18%)、Rh1O5(1h,72%)以及大多數(shù)報(bào)道的電催化劑。相較其他對(duì)照樣,Rh1O4還表現(xiàn)出最高的法拉第效率,并在20輪穩(wěn)定性測(cè)試中還原比例保持95%以上,展現(xiàn)出良好的實(shí)際應(yīng)用前景。EPR、叔丁醇猝滅實(shí)驗(yàn)、H/D動(dòng)力學(xué)同位素實(shí)驗(yàn)等一系列機(jī)理研究證明H*在Rh1O4的4-CP還原過程中起到關(guān)鍵作用,以及Rh1O4上加速的反向氫溢流現(xiàn)象顯著促進(jìn)了還原脫氯反應(yīng)的進(jìn)行。

圖5 鈦基Rh單原子電極4-CP還原活性和脫氯機(jī)理




















所有評(píng)論僅代表網(wǎng)友意見,與本站立場(chǎng)無關(guān)。